
In Vivo ADME: What You Need and Why You Need It
When putting together a data package for regulatory approval by the FDA, EMA, or PMDA, there is a lot to consider and it may be difficult to foresee what studies to include to achieve a ‘good’ pharmacokinetic (PK) profile for a drug candidate. Key to a drug’s entry to first-in-human studies (i.e. a successful Investigational New Drug, or IND, submission) is a data package which includes pharmacology, toxicology, CMC (chemistry, manufacturing, and control), and ADME (absorption, distribution, metabolism, and excretion) properties. A typical IND-enabling ADME package contains data from the following studies: bioanalytical method validation in one rodent and one or more non-rodent species; single- and multiple-dose PK / TK (pharmacokinetics / toxicokinetics); dose proportionality and absolute bioavailability in one rodent and one or more non-rodent species; and in vitro CYP inhibition / induction in human liver microsomes for drug-drug interaction (DDI) assessment (including transporter studies).[1]
Regulatory agencies recommend conducting additional appropriate assays proactively, i.e. prior to IND submission, to avoid late-stage attrition: mass balance and biliary excretion in rats, metabolic profiling and identification (Met ID), and tissue distribution. These components help to form a more complete picture of ADME properties of a compound, and can make an IND package more compelling to get a compound approved for clinical studies. The International Conference on Harmonization recommends the use of “streamlined early exploratory approaches” to gain knowledge of drug candidate characteristics prior to first-in-human studies.[2] Today, more drug developers than ever are heeding this advice and choosing to conduct these studies during preclinical development, and as a result are seeing huge cost savings by reducing need for expensive supplemental studies during Phase I and II of clinical trials.
XenoTech is able to help drug developers plan steps along their non-clinical path and offers a wide array of studies with in vivo ADME experts who have been building experience for nearly 50 years through our partners at the Drug Development Solutions Center in Tokai, Japan.

The Drug Development Solutions Center has conducted countless studies in that time and has been the Japanese industry leader for ADME contract research since 1971. Through high standards and extensive training, the Center offers expert execution of an extensive complement of in vivo ADME / PK services.
An ADME package can be variable depending on a drug candidate’s chemical properties, but a standard in vivo package includes a few common components, outlined below:
Absorption
Pharmacokinetic (PK) study
In a standard PK study, developers are looking to characterize the PK profile of a compound. Drug concentration in plasma is analyzed to characterize the pharmacokinetic properties of the drug. Multiple Sprague-Dawley rats are dosed with radiolabeled compound to provide more complete information about a drug’s pharmacokinetics than a ‘cold’ study yielding only parent compound exposure data. This can be achieved by comparing the concentration of radiolabeled compound in the plasma, representative of both parent and metabolites, to concentration of parent compound. Radiolabeled parent compound is measured using LC-MS, while total reactivity is measured by LSC. If there is a large difference in concentration between the total radioactivity and the parent compound, it indicates that the compound is extensively metabolized. Conversely, if there is almost no difference in concentration between total radioactivity and parent compound, it can be judged that the compound is metabolically stable.
Distribution
QWBA
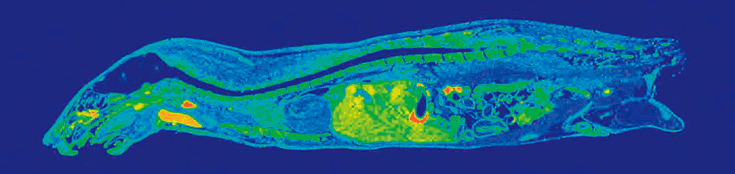
Quantitative Whole Body Autoradiography (QWBA) is a method used to determine a drug’s tissue distribution following administration. For small molecule drugs, usually either a 14C or 3H radioisotope (RI) label is incorporated through RI synthesis to create a radioactive drug analog and is then administered to individuals of a toxicologically-relevant species (typically rat) at different doses. The compound’s distribution is quantified using imaging of a thin slice taken across the entire body at seven time points over 35 days. QWBA may also be performed using pigmented rats for comparison if there is melanin-binding of the compound, which could influence how the drug is localized and terminal half-life. The techniques used to prepare the samples requires extraordinarily high levels of precision and skill, and the Drug Development Solutions Center is unique amongst an already-small number of labs offering the service due to its reputation for consistent, high quality slides and unmatched radiolabeling expertise. In addition to a QWBA study, supplemental tissue dissection is available for consideration of distribution within a particular organ. Radioactivity is a highly sensitive way to quantitatively assess localization of a parent molecule or its metabolites; the value of a QWBA image is that it gives a whole-body picture of distribution and can provide valuable context for toxicology findings and dosimetry data used to inform exposure prediction.
Metabolism
Metabolite ID / profiling
As a drug is metabolized, formation of metabolites can have unintended pharmacological or chemical effects on the body so developers must identify all likely metabolites and understand how they could behave in a patient. Met ID and profiling studies are used to determine how many and what metabolites are formed and the percent of parent exposure (AUC); a metabolite of greater than ten percent of parent AUC would be considered significant and possibly warrant further investigation. These data tie in with in vitro species comparison in microsomes or hepatocytes to ensure there aren’t human-specific metabolites formed, which could be harmful and difficult to explore within test systems. Metabolite profiling and identification are conducted using plasma collected during the PK study, and in urine, feces, and bile collected during the mass balance and biliary excretion studies. Samples are analyzed using HPLC-RAD to ‘profile’ metabolites according to their retention time, and ‘identified’ using LC/MS mass transition.
Excretion
Mass balance
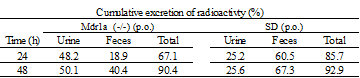
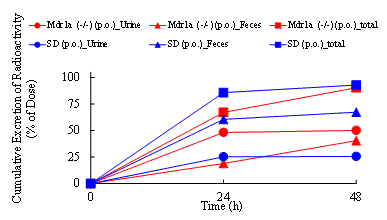
Mass balance studies use radiolabeled compounds to characterize a drug’s excretion path and rate. From this study, the final fate of a compound is obtained. Urine (plus cage wash), feces, in some cases expired air, and the carcass are analyzed for radioactivity to provide a complete picture of how a compound is eliminated from the body and at what rate. The Drug Development Solutions Center has extensive experience with toxicologically-relevant species including rat (albino and Long-Evans pigmented rat), mouse, dog, monkey, rabbit, minipig, and guinea pig, and offers a wide variety of dosing route capabilities. Measurement is usually conducted out to seven days from administration but can be extended through almost complete excretion (more than 95% of dose).
Biliary excretion
Biliary Excretion studies use radiolabeled compounds to evaluate the excretion rate through the bile. From this study, bile-duct cannulated animals are used and bile, urine, and feces are collected for analysis by LC/MS. The Drug Development Solutions Center has extensive experience with the delicacy and surgical skill necessary to perform this type of study and can work not only with rodents, but also large animals such as dog and monkey. Measurement is usually completed after about 48 hours, but timing can be extended depending on the characteristics of the compound.
While the above options outline the standard components of an in vivo ADME package, study components can be tailored to meet the specific needs of the client and extended capabilities are available; ask your XenoTech Services Representative what additional components could be useful to strengthen your drug’s candidacy for regulatory approval.
All RI-labeled compound shipping and handling is seamlessly managed by a designated study director or business development representative who will coordinate all communications throughout the process. Working with our team, clients can be confident they will get signature, unparalleled customer service and technical expertise from XenoTech and the Drug Development Solutions Center. Our teams of experts have built a strong reputation for quick response time and dedicated, experienced study directors handling each and every study, whether carried out in Kansas City or Tokai.
Read more about our in vitro and in vivo study offerings or get in touch with a representative today:
Learn more about:
[1] Wan, Hong. “What ADME Tests Should Be Conducted for Preclinical Studies?” Admet & Dmpk, vol. 1, no. 3, 2013, doi:10.5599/admet.1.3.9.
[2] ICH M3(R2) Guideline: Nonclinical Safety Studies for the Conduct of Human Clinical Trials and Marketing Authorization for Pharmaceuticals; June 2009
Subscribe to our Newsletter
Stay up to date with our news, events and research

Do you have a question or a request for upcoming blog content?
We love to get your feedback
