
Transporters of Emerging Importance in Drug Development: Beyond the Guidance Documents
Dr. Ogilvie’s presentation discusses critical literature and case studies which have been published following the FDA’s 2017 guidance revision, and covered the following topics:
- Regulatory overview of transporters
- Extended clearance classification system (ECCS) to predict rate-determining step in ADCE
- Evidence for emerging transporters – OCT1, OATP2B1, and OAT2
- Unusual routes of administration
Regulatory overview of transporters
The current regulatory view of in vitro transporter studies is detailed in the most recent International Transporter Consortium (ITC) paper from 2018 [1], which notes particular transporters required by FDA, EMA, and PMDA. For example, the FDA and PMDA require compounds to be tested for substrate potential of PgP and BCRP transporters in intestinal epithelia for essentially all orally administered drugs.
The ITC recommends that the choice of transporters to be investigated should be driven by scientific evidence, and should be evaluated either prospectively or retrospectively as described in published literature or guidance. For example, hepatic OCT1 is recommended for prospective evaluation for inhibition and substrate potential, and intestinal OAT2B1 is recommended for retrospective evaluations in specific instances of drug-drug interactions (DDIs). OAT2 is recognized to be an important transporter in the uptake of some high permeability acidic and zwitterionic drugs (ECCS Class 1A), but evidence is lacking for a specific recommendation so drug developers are encouraged to “follow the evidence” to plan studies.
The timing of in vitro studies is also discussed. To plan transporter studies it is best to work backwards from FDA clinical guidance since timing is not explicitly mentioned in the in vitro guidance from 2017. The guidance says that before a drug is to be administered to patients in clinical trials, there should be “enough DDI information to prevent patients form being unnecessarily excluded,” which would come into phase II or later programs, or when a drug is used in oncology or other indications for which patients are enrolled in Phase I.
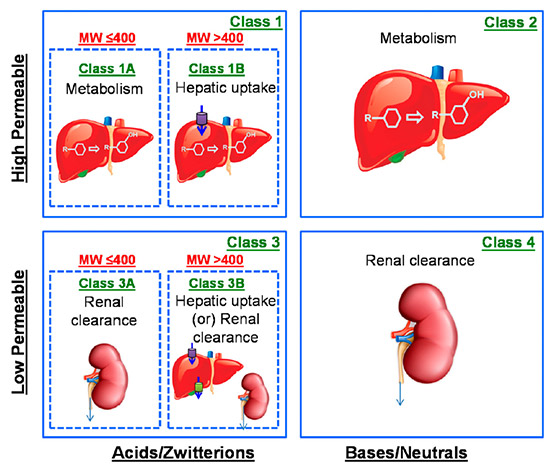
In order to earlier predict the rate-determining step in ADCE (absorption, distribution, clearance, and elimination), the Extended Clearance Classification System (ECCS) was developed and discussed in 2015[2] and 2018 [3] publications in DMD by El-Kattan and Varma. Using this system, compounds can be divided into classes and subclasses based on permeability, ionization, and molecular weight, and each class corresponds to one or more rate-determining steps: metabolism, renal clearance, or hepatic uptake. These rate-determining steps can help predict if transporters or metabolism are more likely to limit the speed of reactions and can help prioritize investigations.
Emerging transporters
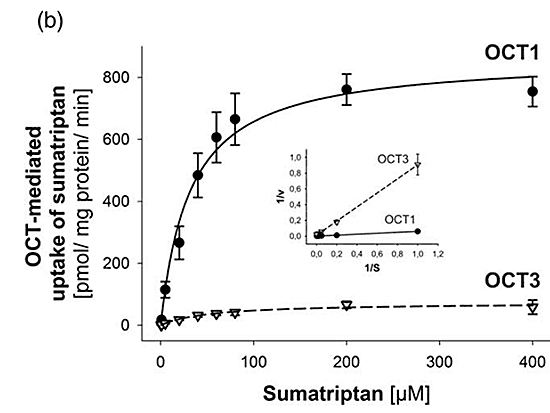
One major transporter discussed is OCT1, the most highly expressed OCT transporter in human liver, located in sinusoidal cells. It is highly polymorphic and transports weak bases, such as metformin, fenoterol, and sumatriptan.
A recent study was published in Clinical Pharmacology & Therapeutics exploring OCT-mediated effects on hepatic uptake and pharmacokinetics of sumatriptan. [4] Sumatriptan is the most commonly prescribed triptan drug on the market, with low bioavailability (<15%) and predominantly metabolized in the liver by MAO-A. It was found that “OCT1 is a high-capacity transporter of sumatriptan and polymorphisms causing OCT1 deficiency have similar effects on sumatriptan pharmacokinetics as those observed in subjects with liver impairment.” There are 5 common polymorphisms in ~9% of Caucasians, which then could mean that those people respond differently to sumatriptan and other triptans than what was clinically predicted, with respect to OCT1 activity. It is observed that the polymorphisms lead to a strong or nearly complete decrease in activity of OCT1 (depending on genotype), reducing clearance and therefore increasing exposure in sumatriptan and other drugs such as tropisetron and morphine (when codeine is administered as the prodrug).
Another important transporter is OATP2B1, which is the most highly expressed OATP in human intestine. There are several noted polymorphisms which haven’t been well categorized yet, but can increase exposure of rosuvastatin and (s)-fexofenadine, and decrease exposure in celiprolol and fexofenadine. There are also complex DDIs associated with OATP2B1, with increases and decreases in exposure of some concomitant drugs depending on whether the OATP transporters involved are intestinal or hepatic. If a drug candidate is a strong OATP1B1/3 inhibitor in a definitive study, inhibition with OATB2B1 should be explored due to high substrate and inhibitor overlap in this family of transporters.[5]
OAT2 is an organic ionic transporter involved in the hepatic clearance of anionic and zwitterionic drugs. OAT2 is the most highly expressed OAT in human liver and is sinusoidal, and is also found to be basolateral in kidney. Transport function is permeability-limited; only low molecular weight acids and zwitterions, classified as 1A drugs according to ECSS, are transported by OAT2.[6] Several very commonly prescribed drugs fall into this category, such as S- and R-warfarin, tolbutamide, and ibuprofen. More instances of drug-drug interactions (DDIs) are expected to come to light, as there is evidence of enzyme-transporter interplay with CYP2C enzyme inhibitors and substrates. Physiologically based pharmacokinetic (PBPK) models that take into account CYP2C and OAT2 interplay can provide better prediction; for example, when tolbutamide is tested using human liver microsomes, the in vivo results can be under-predicted because the effects of enzyme-transporter interplay are not taken into account.
Unusual routes of administration
Drug-drug interactions, specifically within unusual routes of administration, have been increasingly documented, such as a case studies where the combination of multiple existing nasal sprays resulted in obvious clinical changes in exposure. When considering transporter studies, it is important to know the different transporters expressed in different tissues. Dr. Ogilvie also discusses literature detailing nose-to-brain transport considerations for nasally administered drugs[7] and transporters to include when an ophthalmic indication is involved.[8]
An important thing to remember when developing a drug and planning definitive studies is that while guidance provides a framework, additional studies may need to be performed that are outside the guidance. ITC3 recommendations and ECCS classification should be considered when determining appropriate transporter studies, which can provide evidence to support program choices for transporters.
You can watch the webinar here or click here to register for a PDF copy of the slides. XenoTech partners with The Drug Development Solutions Center in Tokai, Japan, to offer an extensive list of transporters for non-clinical studies and highly experienced study directors who provide expert guidance and support throughout contracted studies. Contact us today to plan your drug transporter studies.
Learn more about:
[1] Zamek-Gliszczynski, Maciej J., et al. “Transporters in Drug Development: 2018 ITC Recommendations for Transporters of Emerging Clinical Importance.” Clinical Pharmacology & Therapeutics, vol. 104, no. 5, 2018, pp. 890–899., doi:10.1002/cpt.1112.
[2] Varma, Manthena V., et al. “Predicting Clearance Mechanism in Drug Discovery: Extended Clearance Classification System (ECCS).” Pharmaceutical Research, vol. 32, no. 12, 2015, pp. 3785–3802., doi:10.1007/s11095-015-1749-4.
[3] El-Kattan, Ayman F., and Manthena V. S. Varma. “Navigating Transporter Sciences in Pharmacokinetics Characterization Using the Extended Clearance Classification System.” Drug Metabolism and Disposition, vol. 46, no. 5, 2018, pp. 729–739., doi:10.1124/dmd.117.080044.
[4] Matthaei, J, et al. “OCT1 Mediates Hepatic Uptake of Sumatriptan and Loss-of-functionOCT1polymorphisms Affect Sumatriptan Pharmacokinetics.” Clinical Pharmacology & Therapeutics, vol. 99, no. 6, 2016, pp. 633–641., doi:10.1002/cpt.317.
[5] UW DIDB e-Pkgene; Zamek-Gliszczynski et al. ITC3, 2018.
[6] Kimoto, Emi, et al. “Organic Anion Transporter 2–Mediated Hepatic Uptake Contributes to the Clearance of High-Permeability–Low-Molecular-Weight Acid and Zwitterion Drugs: Evaluation Using 25 Drugs.” Journal of Pharmacology and Experimental Therapeutics, vol. 367, no. 2, 2018, pp. 322–334., doi:10.1124/jpet.118.252049
[7] Djupesland, Per G, et al. “The Nasal Approach to Delivering Treatment for Brain Diseases: an Anatomic, Physiologic, and Delivery Technology Overview.” Therapeutic Delivery, vol. 5, no. 6, 2014, pp. 709–733., doi:10.4155/tde.14.41.
[8] Lee, J., and R. M. Pelis. “Drug Transport by the Blood-Aqueous Humor Barrier of the Eye.” Drug Metabolism and Disposition, vol. 44, no. 10, 2016, pp. 1675–1681., doi:10.1124/dmd.116.069369
Subscribe to our Newsletter
Stay up to date with our news, events and research

Do you have a question or a request for upcoming blog content?
We love to get your feedback
